Hugh Shanahan |

While microarray data has been extensively employed in understanding Biological systems and on the algorithms to infer interactions, comparatively little has been done in methods to identify biased subsets of micro-array data. In collaboration with Andrew Harrison's team at the University of Essex we have been examining one particular class of biases that have a biophysical origin. Here's a paper we've published in Nucleic Acids Research on this.

As the cost of sequencing has collapsed the amonut of data being generated has exploded. Sequencing centres are now producing hundreds of Tbytes of data. The wide variety of different data sets being analysed (genomes, RNA-seq, ChIP-seq, methylome, anbd so on) must be analysed to determine their quality, the underlying sequence and ultimately the underlying Biology. Cloud computing platforms present an opportunity to circumvent this difficulty by co-hosting the analysis of the data with its generation. At present, we have received from the EU, via the Venus-C collaboration to explore the analysis of transcriptomic (micro-array) data sets using the Microsoft Azure cloud computing platform.
 |
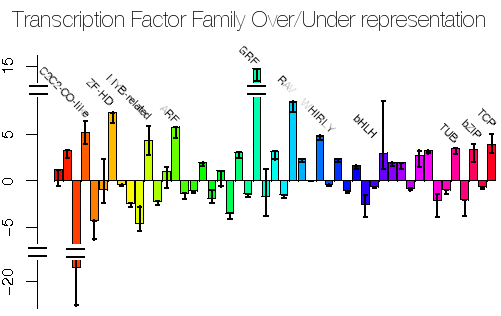 |
Since arriving at Royal Holloway I have been working been working closely with the Plant Science group at Royal Holloway, in particular with Enrique Lopez, on analysing the expression of genes in the early development of Arabidopsis thaliana. I was also an advisor for a Ph.D. studentship in the construction of high-order gene regulatory networks related to plant responses to biotic, abiotic stresses and hormones, supervised by Alessandra Devoto. There is an intense amount of interest in the field of Systems Biology (even though I've yet to see a good definition for it yet) and networks in general and we have founded the Centre for Systems and Synthetic Biology.
| I gave a talk on this in University College Cork, click on the format that suits you: |  |
|
| Here is another, slightly shorter version of this I gave at a meeting in Hangzhou: | |
Recent publications in this area
Distinct Light- Mediated Gene Expression and Cell Cycle Programs in the Shoot Apex and Cotyledons,Enrique Lopez-Juez, Edyta Dillon, Zoltan Magyar, Safina Khan, Saul Hazledine, Sarah M. de Jager, James A.H. Murray, Gerrit T.S. Beemster, Laszlo Bogre, and Hugh Shanahan, Plant Cell, 20 (4), 947-968, (2008).
Alignment-based methods for classifying sequences have been spectaculorly successful in a number of different areas of Bioinformatics. However, there exists a number of different domains, such as the classification of Transcription Factor Bindind Sites where such methods have not been very succesful and different approaches such as motif searches have been employed. A very different approach to this is to use the principle of string kernels based on word frequencies and a Support Vector Machine formalism. To this end, we developed a easy-to-use tool called SKM to make use of this technique. We have demonstrated that this apporach is competitive with one of the best motif-search tools used at present.
You can find our submission for this tool to F1000 here.
Protein-DNA interactions |
 |
 |
DNA-binding proteins play a very important role in the function of all cells. In particular, they are involved in transcription (reading data from DNA and converting into strands of RNA), DNA repair (which reduces enormously the amount of mutations that occur within DNA and is essential for viable reproduction to occur) and transcriptional regulation. There is an intense amount of interest in transcriptional regulation as this is fundamental in cell differentiation (not all genes in complicated organisms such as mammals are active in any particular cell), cell reproduction and in the dynamic response of a cell to stresses e.g. Oxygen shortages. In understanding transcriptional regulatory networks, it is hoped we can derive a much deeper understanding as to how cells function through their life cycle and in the way they respond to immediate stresses. Furthermore, from a Biomedical perspective many Oncogenes, genes that play a role in Cancer, give rise to proteins involved in transcriptional regulation.
I am interested in understanding how transcription factors are able to identify specific binding regions and how one can identify novel protein structures that bind to DNA. In the latter case, I have written a web server that can identify proteins that bind to DNA with a Helix-Turn-Helix motif called HTHquery.
| I gave a talk on this in Glasgow, click on the format that suits you: | |
Publications in this area
HTHquery, a method for detecting
DNA-binding proteins with a Helix-Turn-Helix structural motif,
H.P. Shanahan, C. Ferrer, S. Jones, and J.M.Thornton, Bioinformatics 21 (18) 3679-3680
(2005).
Identifying DNA binding proteins
using structural motifs and the electrostatic potential,
H.P. Shanahan, M.A. Garcia, S. Jones and J.M. Thornton, Nucl Acid Res 32, (16), 4732-4741,
(2004).
Solubility as an evolutionary constraint |
 |
Publications in this area
Amino Acid architecture and the
distribution of polar atoms on the surfaces of proteins,
H.P. Shanahan and J.M. Thornton, Biopolymers, 78, (6), 318-28,
(2005).
An examination of the
conservation of surface patch polarity for proteins,
H.P. Shanahan and J.M. Thornton, Bioinformatics,
20 (14)
2197-2204, (2004)
Zhejiang University (Hangzhou)
Chinese Agricultural University (Beijing)
Huazhong Agricultural University (Wuhan)
We are very interested in running this school elsewhere in the developing world. Please get in touch with us.
| Course Title | Course Code and Web Site | |
| Robotics | CS2830 | |
| Concurrent and Parallel Programming | CS3750 |
